


【建模文章解读】TPN729MA的临床前药代动力学,并通过PBPK模型预测其人体PK

原文献作者
Zhi-wei GAO1, 2, Yun-ting ZHU2, Ming-ming YU2, Bin ZAN2, Jia LIU2, Yi-fan ZHANG2, Xiao-yan CHEN2, Xue-ning LI1, Da-fang ZHONG2
1复旦大学附属中山医院临床药理学教研室
2中国科学院上海药物研究所
导 读
该案例建立了TPN729MA的大鼠和比格犬的PBPK模型,并分析了在其体内的ADME过程,评估了GastroPlus软件內建的计算稳态分布容积的方法,同时采用5种不同的方法推算得到人体的清除率,最终完成了该IND的首次人体PK预测,预测的PK曲线与实测结果的Tmax,Cmax和AUC在2倍误差范围之内。
推荐理由
本文以TPN729MA为例完整展示了首次人体PK预测的常规流程,包括在搭建人体PBPK模型的过程中涉及的体外数据的获取、Vss计算方法的确定、多种异速放大和IVIVE计算人体CL的方法,对于初次接触首次人体PK预测的研发人员具有较好的参考价值。
案例摘要
TPN729MA是目前正在中国临床开发的新型选择性PDE5抑制剂,用于治疗勃起功能障碍。在该研究中,表征了其大鼠和比格犬中药代动力学(PK),使用GastroPlus软件搭建生理的药代动力学(PBPK)模型,预测了TPN729MA的人体PK,在该模型中TPN729MA的人体清除率(CL)是使用临床前种属的清除率通过四种异速放大方法及体外人肝微粒体固有清除率IVIVE转换得到,模型中人体稳态分布容积(Vss)的计算方法是通过使用临床前种属的PK特征评估GastroPlus內建的不同Vss的计算方法确定,最终模型中采用的人体Vss的计算方法同样为Rodgers和Rowland开发的方法。考虑到预测的不确定性,本研究还对3名健康男性受试者进行了初步的人体研究,研究结果表明实测结果与预测的PK曲线相似,并且预测的Tmax,Cmax和AUC值均在实测值的2倍误差范围之内。
软件用途
本案例中使用体外实验数据、整合GastroPlus软件內建的临床前动物种属的胃肠道ACAT模型和生理学参数,搭建了大鼠和比格犬的PBPK模型;进行首次人体PK预测时,采用了软件內建的人体胃肠道ACAT模型、生理学参数、Vss计算方法,结合基于异速放大及IVIVE计算得到的清除率搭建了人体PBPK模型,最后完成了首次人体PK预测。
1. 研究背景
勃起功能障碍(ED)对生活质量产生明显的负面影响。实现勃起的生理机制是通过一氧化氮-环鸟苷单磷酸(cGMP)途径介导。磷酸二酯酶5型(PDE5)抑制剂可抑制动脉壁平滑肌细胞中cGMP的磷酸二酯键的降解,通过维持在海绵体和供应它的血管细胞中足够的cGMP水平来增强勃起功能。
西地那非是第一种用于治疗ED的口服PDE5抑制剂,并于1998年获得美国FDA批准。此后,新的PDE5抑制剂(例如他达拉非、伐地那非和阿伐那非)已在世界范围内被批准,乌地那非和米罗那非在韩国批准用于治疗ED。
TPN729MA是一种新型的选择性PDE5抑制剂(结构式见图1),用于治疗ED。天方药业从2015年开始TPN729MA项目(上海特化医药开发)的临床试验,2019年处于II期临床试验中。与其他已知的磷酸二酯酶相比,对PDE5的选择性高。
本文发表于2015年,当时还未开展I期临床试验。在开展I期临床之前,采用临床前数据预测了首次人体PK,为临床试验的开展与设计提供指导。
TPN729MA对PDE5的中位抑制浓度(IC50)为2.28 nmol / L,与他达拉非(2.35 nmol / L)相似,且低于西地那非(5.22 nmol / L)。此外,在大鼠和狗的勃起模型中,与安慰剂相比,TPN729MA显着增加了最大海绵体内压(ICP)和ICP与血压的比率。
作者研究了TPN729MA在大鼠、比格犬口服和静脉内给药后的PK特性;及体外特性:Caco-2细胞渗透性、血浆蛋白结合率、全血-血浆分配系数Rbp和肝微粒体代谢稳定性。基于体外和动物体内试验获得的临床前数据,通过PBPK模型对TPN729MA的动物和人体PK曲线进行模拟。预测了TPN729MA的人体PK曲线,以评估其临床PK和进一步临床开发的风险收益比评估。
2. 建模数据与处理
2.1 TPN729MA的相关建模参数
表1-TPN729MA理化和生物药剂学性质及清除参数

2.2 数据获取及处理
为准确地预测TPN729MA的口服吸收速率和程度,该项目使用了高级房室吸收转运模型(ACAT模型),ACAT模型是在Yu和Amidon的房室吸收转运模型CAT基础上开发而来。模拟需要输入的主要参数包括:分子量、pKa、logP、溶解度、Caco-2渗透性、大鼠,比格犬和人的全血-血浆分配系数Rbp、游离药物分数fu和清除率CL值(表1)。大鼠和比格犬静脉和口服PK数据(表3和图2)均通过试验测量得到,并对3位健康中国男性受试者进行了初步人体PK研究,口服剂量为25 mg。
预测人体PK时,假定组织和血液之间的药物分配受到灌注速率的限制,肝脏和肾脏作为清除器官。将模拟的PK参数与实测的数据进行比较,以评估预测的准确性。
非房室分析:静脉给药后,全身血浆清除率(CL)计算方法为CL = Dose /AUC0-∞;稳态分布体积(Vss)计算方法为Vss = CL×MRT;口服绝对生物利用度(F)计算方法为F =(Doseiv / Dosepo)×(AUCpo / AUCiv)×100%。
表3-TPN729MA静脉推注和口服后在大鼠和比格犬中的药代动力学参数
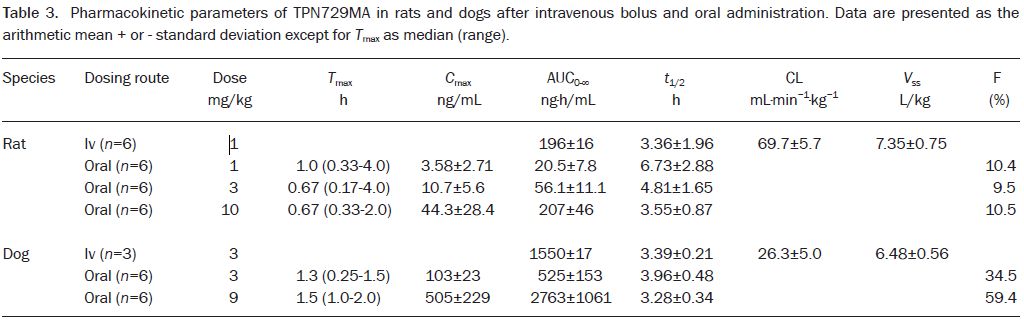
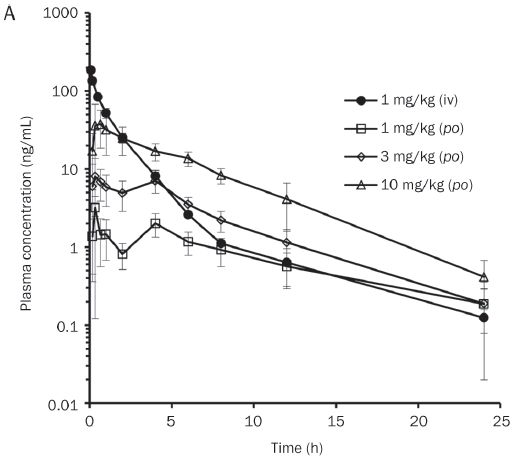
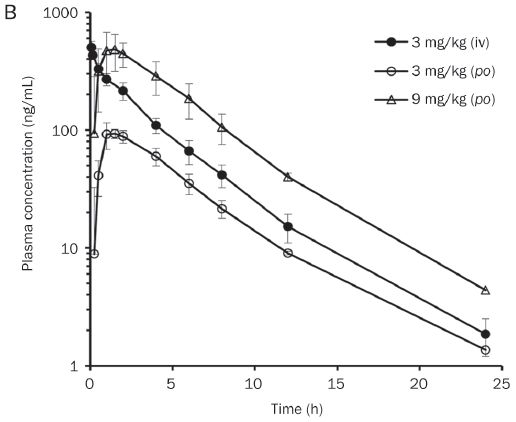
图2. 静脉(iv)或口服(po)TPN729MA后,大鼠(A)和比格犬(B)中TPN729MA的PK曲线。 在大鼠中剂量为1 mg/kg(iv),1,3和10 mg/kg(po)。 比格犬的剂量分别为3 mg/kg(iv)和3,9 mg/kg(po)。
人体组织血浆分配系数(Kp值)的获取及处理:使用基于组织成分的方法估算Kp值。 GastroPlus 8.6中提供了两种已公开的机制方法,可使用理化和体外数据(例如logP和fu)来预测Kp值。其中方法一:基于Poulin和Theil 的方法,并由Berezhkovskiy 进行了校正;方法二:基于Rodgers和Rowland开发的方法。使用不同的Kp值计算方程式预测的TPN729MA的Vss值在表4中显示。与其他方法相比,Rodgers和Rowland开发的方程式计算得到的大鼠和比格犬的Vss最准确,使用该Vss值和静脉给药计算得到的CL作为输入值,对大鼠和比格犬的静脉给药后的实测PK曲线可以实现较准确的预测(图3)。据此,人体PBPK模拟同样选择了Rodgers和Rowland开发的Vss的估算方法。
表4-使用不同方程对大鼠、比格犬和人体中TPN729MA 的Vss预测结果
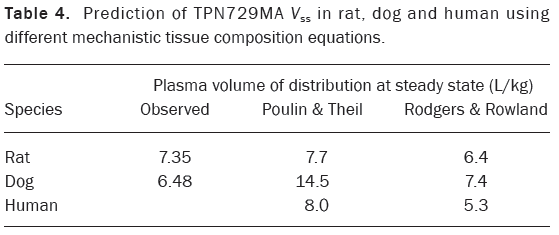
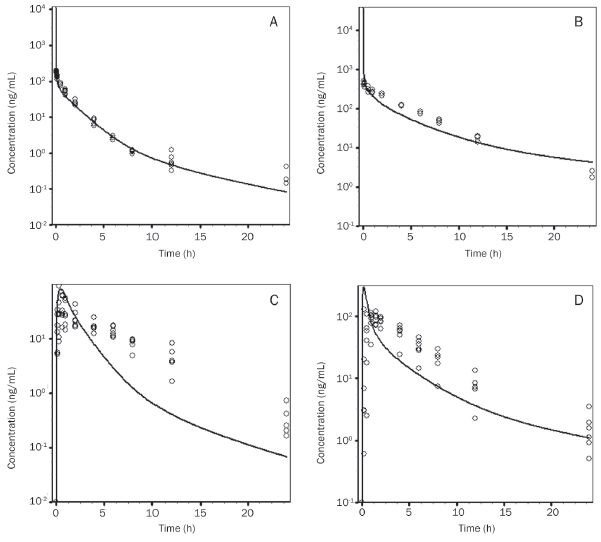

图3. 大鼠(A)或比格犬(B)静脉给药和大鼠(C)或比格犬(D)口服给药后,TPN729MA的实测值和PBPK模型预测的PK曲线。 空心圆圈表示从单个动物实测数据,实线表示根据GastroPlus中的PBPK模型预测的PK曲线。 大鼠的给药剂量为1 mg/kg(iv)和10 mg/kg(po),比格犬的给药剂量为3 mg/kg(iv)和3 mg/kg(po)。
清除率的获取及处理:大鼠和比格犬PBPK模型中对应的清除率是通过单次静脉注射TPN729MA获得的体内PK曲线计算得到,人体PBPK模型中清除率的计算采用了两类方法:体外-体内外推(IVIVE)和异速放大法。
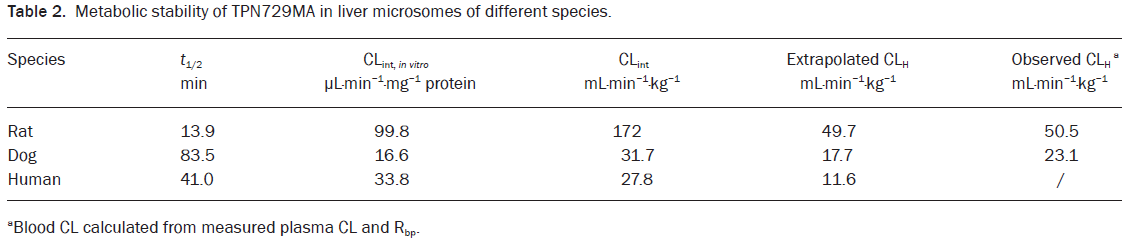
IVIVE法:通过体外肝微粒体代谢稳定性试验计算得到肝脏清除率(表2),具体步骤为:Step1: 根据体外肝微粒体代谢稳定性试验中TPN729MA消除的t1/2计算出体外固有清除率CLint,in vitro。
CLint,in vitro =(0.693 / t1/2)×(1 / Cprotein)
其中Cprotein是孵育过程中的蛋白浓度, t1/2由浓度与时间曲线的对数线性回归分析的斜率(k)确定,t1/2= ln2 / k。
Step2:使用基于生理的比例因子计算得到体内清除率CLint
CLint =CLint,in vitro×(mg protein / g liver weight)×(g liver weight / kg body weight)
其中大鼠、比格犬和人的mg protein / g liver weight分别为47、58和32;大鼠、比格犬和人的g liver weight / kg body weight分别为36.6、32.9和25.7。
Step3:使用CLint和肝血流量Q计算得到体内肝脏清除率(CLH),在充分搅拌肝脏模型中,不考虑药物的结合,CLH的计算方法为:
CLH =(Q × CLint)/(Q + CLint), 肝提取率为CLint / Q。
其中大鼠、比格犬和人的肝血流量Q分别为70、40和20 mL·min-1·kg-1)
表2-TPN729MA代谢稳定性
异速放大法(结果见表1):
方法一:基于大鼠异速放大(SSSrat);
方法二:基于比格犬异速放大(SSSdog)
CLu, human = CLu, animal × (BWhuman / BWanimal) 0.75, 其中大鼠、比格犬和人的体重分别为0.22、8.4和 70kg,CLu表示血浆中游离药物的清除率。
方法三:使用游离分数截距校正法(FCIM)预测CL
CLhuman = 33.35 mL / min (a / Rfu ) 0.77
其中,a是从大鼠和比格犬的对数-对数图的截距中获得,Rfu是药物在大鼠与人血浆中的fu之比。
方法四:使用两种物种缩放方法(TSrat-dog)预测CL的水平
CLhuman = a (rat-dog) × BWhuman0.628
其中a (rat-dog) 是从大鼠和比格犬的对数-对数图的截距中获得。
3. 结果与分析
TPN729MA在大鼠、比格犬和人的体外肝微粒体的固有清除分别为99.8、16.6和33.8μL·min-1·mg-1 protein,代谢速率遵循大鼠>人>狗,在大鼠、比格犬和人体内固有清除率分别为172、31.8和27.8mL·min-1·kg-1,外推得到肝脏清除分别为49.7、16.7和11.6 mL·min-1·kg-1,准确地预测了大鼠和比格犬体内清除率(1.02倍和1.31倍)(表2)。
大鼠中以1 mg/kg的剂量和比格犬以3 mg/kg的剂量单次静脉注射TPN729MA,总血浆清除率(CLp)分别为69.7和26.3 mL·min-1·kg-1。根据每种物种的体外Rbp值(1.38和1.14),大鼠和比格犬的血液清除率(CLb)分别为50.5和23.1 mL·min-1·kg-1。大鼠和比格犬的肝提取率(CLb / QH)分别为72.1%和57.8%,表明TPN729MA在临床前动物中显示出中等偏高的清除率。在大鼠和比格犬中,稳态分布分别为7.35和6.48 L/kg,这表明其广泛分布到组织中(> 9倍于全身水)。在大鼠中分别以1、3和10 mg/kg口服TPN729MA后,在给药后0.67-1小时,TPN729MA的Cmax值为3.58、10.7和44.3 ng/mL,AUC值分别为20.5、56.1和207ng·h/ mL,Cmax和AUC的增加大致与剂量成比例,T1/2为3.55-6.73 h,口服绝对生物利用度约为10%。在比格犬中以3和9 mg / kg的剂量单次口服TPN729MA后,在给药后1.4小时达到TPN729MA的Cmax,AUC值分别为525和2763ng·h/ mL,Cmax和AUC的增加大于剂量比例,T1/2为3.6小时,绝对生物利用度分别为34.5%和59.4%(表3和图2)。
人体PK的预测,使用临床前种属(大鼠和比格犬)实测的Vss评估GastroPlus软件中内建的Kp估算方法,最终发现使用Rodgers和Rowland开发的方程式估算的大鼠和比格犬中Vss的最准确。使用该Vss值和静脉给药计算得到的CL作为输入值,对大鼠和比格犬的静脉给药后的PK曲线可以实现较准确的预测,同时将溶解度数据输入到GastroPlus中的可以很好地模拟口服给药后的PK曲线(图3),TPN729MA在大鼠和比格犬中的预测AUC值分别是实测值的1.4倍和1.6倍,因此人体Kp值的估算同样采用了Rodgers和Rowland开发的方程式。使用在人肝微粒体中测定的体外固有清除率CLint,估算得到人体血浆清除率(CLp)为12.3 mL·min-1·kg-1。使用SSSrat,SSSdog,FCIM和TSrat-dog方法估算得到人体CLp分别为10.3、11.1、6.42和9.53 mL·min-1·kg-1。除FCIM外,使用其他预测方法预测的人类CL没有明显差异(在1.3倍以内)。
在尚不清楚哪个CL值与首次人体PK研究实测值最为接近时,将这些CL估算值都与PBPK模型相结合,以预测TPN729MA的人体PK曲线。根据体表面积,从大鼠有效剂量2.5 mg/kg推算出TPN729MA人体(70kg)的有效剂量约为25mg。
模拟人体口服25 mg TPN729MA后的PK,预测结果和实测的人体PK参数以及PK曲线,如图4和表5所示。总体PBPK模型合理地拟合了人体口服25mg TPN729MA的PK曲线,使用TSrat-dog,SSSrat和SSSdog方法估算的CL预测的PK参数(Tmax,Cmax,AUC和T1/2)均在实测值的2倍误差以内,而使用FCIM估算的CL 预测的TPN729MA血浆暴露(AUC)高估(2倍误差),使用HLM估算的CL 预测的血浆暴露低估(1.9倍误差)(表5)。
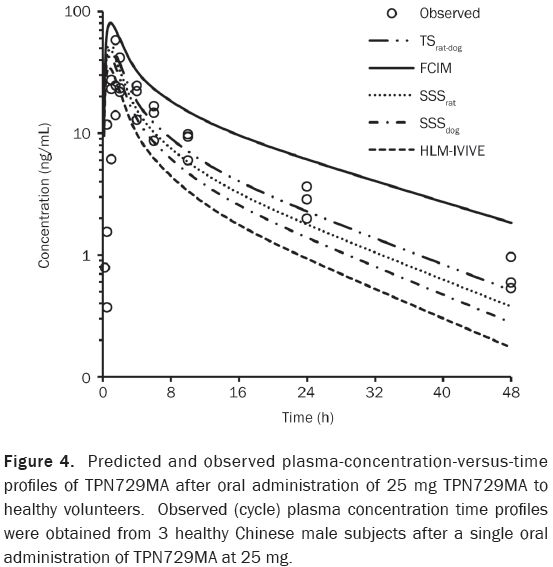
图4. 3名中国健康男性受试者口服25 mg TPN729MA预测和实测PK曲线。
表5. 人体口服25 mg TPN729MA后的预测和实测的PK参数。括号中的倍数误差表示预测的准确性(预测值与实测值的比或实测值与预测值的比)。

4. 讨论
TPN729MA是用于治疗ED的新型选择性PDE5抑制剂,2015年文章发表时处于临床开发阶段。TPN729MA具有高溶解度(在水中为30 mg / mL)、中等亲脂性的弱碱。 TPN729MA在Caco-2渗透试验中表现出中等偏高的渗透性,结合高溶解度的特性其可能在人体中表现出较高的肠道吸收率。
TPN729MA在临床前动物中显示中等偏高清除率。通过使用各种属的Rbp,将大鼠和比格犬的系统血浆CL转换为血液CL,大鼠的CL(占肝血流量的72%)高于狗的CL(占肝血流量的58%)。口服给药后大鼠的较低暴露与静脉内给药后观察到的较高的CL一致。当使用体外肝微粒体CL来预测大鼠和比格犬的体内CL时,预测值和实测CL值之间具有极好的相关性(大鼠为1.02倍,狗为1.31倍),基于这种相关性以及在大鼠和比格犬中均未低估系统CL的事实,肝脏代谢可能是临床前种属清除的主要途径。通过大鼠口服TPN729MA的质量平衡研究表明,通过尿液和胆汁消除母体药物的作用微不足道(小于剂量的1%,数据未显示),从而证实TPN729MA是通过肝脏代谢清除。
有关PBPK方法在预测人体药代动力学方面的实用性的报告有很多。比如商用PBPK软件GastroPlus(http://www.simulations-plus.com)越来越多地用于制药领域中人体PK的回顾或前瞻性预测。使用PBPK模型模拟人体PK曲线,必须估算化合物的CL和分布。另外,口服PK模拟的重要组成部分是预测药物的吸收速率和吸收程度。本研究通过临床前种属的体内数据结合体外数据,预测了人体口服TPN729MA的PK。在PBPK模型的搭建过程中,首先在大鼠和比格犬中验证了整个PBPK模型,该模型合理地拟合了TPN729MA在大鼠和比格犬经静脉和口服给药后的PK曲线,AUC预测误差在实测值的1.6倍以内,根据文献,PBPK建模预测误差小于两倍被认为是准确的。
在本文中,使用不同的方法预测了TPN729MA的人体CL,包括IVIVE,SSS以及PhMRA CPCC推荐的两种预测方法:FCIM和TSrat-dog。通过查阅文献,回顾对CL预测方法发现,如果CL主要是由CYP450代谢,则使用人肝微粒体估算的CL应与SSS一致,在本研究中,使用人肝微粒体预测的体内CL(12.3 mL·min-1·kg-1)与SSSrat(10.3 mL·min-1·kg-1)、SSSdog( 11.1 mL·min-1·kg-1)预测的CL一致。根据当前研究获得信息,假定TPN729MA主要通过人肝脏代谢清除,基于低CL(6.42 mL·min-1·kg-1)至高CL(12.3 mL·min-1·kg-1),TPN729MA的CL在人肝血流中分别占32%至62%。由于TPN729MA的人体PK曲线预测具有不确定性,进行了一项初步的人体研究,口服剂量为25 mg,用于准确评估健康男性受试者的PK。体外数据结合GastroPlus软件中內建的ACAT模型,预测的口服25 mg TPN729MA后的Cmax为实测值的1.0到2.2倍,预测的AUC是实测值的1.0到2.0倍。通常,很难前瞻性地知道哪种预测方法最适合新化合物,在这项研究中,TSrat-dog方法在人体PK预测中提供了最准确的预测(AUC为1.0倍,Cmax为1.5倍)。 SSSrat和SSSdog方法提供了相似的预测精度。FCIM和IVIVE的预测准确性比TSrat-dog,SSSrat和SSSdog的预测准确性差。
总的来说,TPN729MA在大鼠和比格犬中的PK特征是吸收迅速,中高CL,高分布和低中度生物利用度。通过使用多种体外和体内预测方法预测人体CL,结合PBPK模型预测了TPN729MA的人PK曲线。本研究的局限性是相对较小的临床样本量,在未来的临床开发过程中,可以对该PBPK模型进行完善,以整合现有临床研究中有关药物处置的其他信息。成功预测人体PK曲线会使药物开发过程中化合物的损耗减少,降低由于临床试验设计失败导致的时间和成本损失。该PBPK模型可以继续扩展以预测较宽的剂量范围,促进临床研究和剂量递增的临床试验设计,并探索食物的作用和可能的药物相互作用。
5. 总结
该研究搭建了TPN729MA的大鼠和比格犬PBPK模型,描述了其大鼠和比格犬药代动力学的特征,使用临床前种属的PK特征评估了GastroPlus內建的不同Vss的计算方法,并选择使用Rodgers和Rowland开发的方法计算人体Vss,使用四种异速放大方法及IVIVE转换得到人体CL,最终完成了首次人体PK预测,预测的PK曲线与实测结果的Tmax,Cmax和AUC在 2倍误差范围之内。
6. 软件应用
该案例应用的软件是GastroPlus (version 8.6),涉及模块有Base, PBPK.
参考文献
Zhi-wei GAO1, 2, Yun-ting ZHU2, Ming-ming YU2, Bin ZAN2, Jia LIU2, Yi-fan ZHANG2, Xiao-yan CHEN2, Xue-ning LI1, Da-fang ZHONG2. Preclinical pharmacokinetics of TPN729MA, a novel PDE5 inhibitor, and prediction of its human pharmacokinetics using a PBPK model. Acta Pharmacologica Sinica (2015) 36: 1–9 IF:4.01