


【建模文章解读】通过体外研究、静态机制模型和PBPK模型评估促变药Esaxerenone的药物相互作用风险
原文作者
Makiko Yamada, Tomoko Ishizuka, Shin-ichi Inoue, Veronika Rozehnal, Thomas Fischer,and Daisuke Sugiyama
1. 第一三共(日本)制药公司,药物代谢和药代动力学研究实验室;
2. 第一三共(欧洲)制药公司,组织与细胞研究中心(慕尼黑)。
解读人
王钰玺,凡默谷技术部
导 读
本研究通过评估体外、静态机制模型和PBPK模型评估了小分子药物Esaxerenone作为促变药产生DDI的风险。静态机制模型分析表明CYP3A介导的Esaxerenone的潜在DDI不可忽略。PBPK模型模拟结果表明由于TDI作用和诱导作用的抵消效应,该药物作为促变药产生DDI的可能性较低,模拟结果与已有的临床研究结果相近。
推荐理由
在本研究中使用体外数据分析了不同酶和转运体介导产生DDI的可能性,结果表明Esaxerenone作为的CYP3A的TDI和诱导剂,以及2B6的抑制剂和诱导剂需要进一步模拟分析DDI风险。接着通过静态机制模型评估了CYP3A和2B6介导的DDI,结果表明CYP3A介导的潜在DDI不可忽略。考虑到静态机制的DDI模拟可能会造成假阳性,又通过PBPK模型进一步评估了DDI风险,结果表明由于抑制作用和诱导作用的抵消效应,Esaxerenone作为促变药产生DDI的可能性较低。并且在构建PBPK模型时还考虑了不同研发阶段的建模数据的完整性,因此从学习DDI研究的整体流程以及结合实际研发进展的PBPK建模方面都具有重要的参考意义。
案例摘要
Esaxerenone(CS-3150)是一种新型的口服非甾体选择性盐皮质激素受体阻滞剂,已在日本批准用于治疗高血压。本研究通过体外研究评估了Esaxerenon药物-药物相互作用(DDI)的可能性。Esaxerenone是CYP3A的时间依赖性抑制剂(TDI)和诱导剂,使用静态机制模型分别评估了Esaxerenone对CYP3A抑制和诱导的临床影响,结果表明咪达唑仑(典型的CYP3A底物)曲线下面积的比率(AUCR)分别为1.80和0.31,因此Esaxerenone的潜在DDI不可忽略,且DDI可能主要发生在肠道中。考虑到静态机制模型可能会高估预测结果,又使用PBPK模型做了进一步评估,并预测了胃肠道中的浓度-时间曲线。尽管同时具有抑制作用和诱导作用的化合物的DDI难以预测,但使用动态PBPK模型预测的咪达唑仑AUCR约为1.2,与临床研究结果相近。本研究还模拟了其他临床用药场景,结果表明当仅考虑抑制或诱导作用时,咪达唑仑的AUCR会随Esaxerenone的给药时间、给药剂量、以及咪达唑仑的给药时间而改变。但是,综合考虑两种作用时模拟的AUCR几乎保持不变。表明由于抑制作用和诱导作用的抵消效应,Esaxerenone作为促变药产生DDI的可能性较低。
软件用途
在本案例中使用GastroPlus搭建了三个PBPK模型,以考察药物开发不同阶段、不同完整性的建模数据使用GastroPlus搭建的模型对DDI的预测性。模拟了肠道中的药物浓度、讨论了GastroPlus用于预测肠道中弱DDI的实用性。还模拟了新的用药场景,如Esaxerenone与咪达唑仑不同给药时间的DDI,以及不同剂量的Esaxerenone与咪达唑仑的DDI。
正文目录
1. 研究背景
Esaxerenone(CS-3150)是一种新型的口服非甾体类选择性盐皮质激素受体阻滞剂,已于2019年1月在日本批准用于治疗高血压。目前正在研究用于治疗糖尿病肾病。由于其高膜渗透性,Esaxerenone在口服后迅速吸收,具有较高的生物利用度。尽管它的主要消除途径是代谢,但由于它通过多种途径代谢,例如氧化、水解和葡糖醛酸化,因此认为Esaxerenone作为受变药与酶抑制剂发生药物-药物相互作用(DDI)的程度有限。为了保证用药安全,FDA、EMA、日本厚生省等法规部门都要求对新药进行体外DDI的相关研究。当体外研究表明某种药物对代谢酶或转运体具有抑制或诱导能力时,通常首先使用静态模型估算该药物在临床环境中的作用。然而,由于在使用静态模型时假设了高药物浓度,往往会导致DDI程度的高估。因此,基于生理的药代动力学(PBPK)模型被认为是评估DDI的更接近真实世界的预测方法。在提交给FDA的与DDI有关的PBPK模型中在研药物作为促变药相比受变药的案例较少。对于在体外同时显示抑制和诱导作用的化合物,FDA的DDI指南建议体内抑制和诱导作用分别模拟,因为同时预测可能会导致假阴性结果。此外,当预测CYP3A介导的弱DDI时,肠道中酶活性的变化对体内相互作用起主导作用而不是肝脏,因为肠细胞中的药物浓度通常高于肝细胞中的药物浓度。先前的大多数研究都使用强抑制剂PBPK模型模拟CYP3A介导的口服药物DDI,肠道CYP3A被完全抑制,而对CYP3A介导的弱DDI的预测性能还需要进一步评估。在本研究中,通过体外研究评估了Esaxerenone对CYP450亚型,UGT1A1,UGT2B7和几种转运蛋白的抑制特性,以及对CYP1A2,CYP2B6和CYP3A4的诱导作用。体外评估结果表明无法忽略临床DDI风险,进一步使用了静态机制模型和PBPK模型进行了DDI风险评估。由于制药公司从药物发现阶段就会引入PBPK模型,只有动物和体外数据可用,因此我们比较了有和没有人体PK数据时模型对DDI的预测准确性。通过PBPK模型模拟了Esaxerenone不同给药周期、不同剂量、咪达唑仑的不同服用时间对DDI的影响,讨论了抑制和诱导之间的抵消效应。
2.研究方法
2.1 体外DDI研究
1) 使用人肝微粒体研究了Esaxerenone对CYP1A2,CYP2A6,CYP2B6,CYP2C8,CYP2C9,CYP2C19,CYP2D6,CYP2E1和CYP3A4的可逆抑制和时间依赖性抑制(TDI)。Esaxerenone是UGT亚型的底物,使用人肝微粒体研究了Esaxerenone对UGT1A1和UGT2B7的可逆抑制的潜力。
2)考察了Esaxerenone对下列蛋白的抑制作用:转染的MDCK-II细胞中人有机阴离子转运蛋白OAT1和OAT3、有机阴离子转运多肽OATP1B1和OATP1B3、有机阳离子转运蛋白OCT1和OCT2、转染的HEK293细胞中人多药及毒素外排转运蛋白MATE1和MATE2-K、Caco-2细胞中的人乳腺癌耐药蛋白BCRP和p-糖蛋白(P-gp)。
2.2 使用静态机制模型的DDI风险评估
根据FDA的DDI的指导原则(2020年),药物在肠腔内的浓度(Igut)=[剂量] / [250 ml] = 42.9 mM。如果Igut/ IC50小于10,则认为Esaxerenone在体内抑制P-gp和BCRP转运蛋白的能力较低很。使用静态机制模型评估了Esaxerenone作为CYP3A的抑制剂和CYP2B6的诱导剂对咪达唑仑AUC的影响,计算公式如下:

其中,Fg:被小肠吸收且未经肠代谢的药物分数;fm:在总体肝脏清除中受抑制剂或者诱导影响的CYP酶介导的底物清除分数。下标 h表示肝脏;g表示肠道。假设咪达唑仑吸收分数(Fa)为1,根据咪达唑仑的绝对生物利用度(0.30)和通过肝的量(0.56)计算得出咪达唑仑的Fg为0.54。将咪达唑仑的fm设置为0.94,将CYP2B6底物的fm设置为1(以评估最大DDI风险)。上式中A,B和C分别表示可逆抑制、TDI和诱导作用,并分别根据等式2-4计算:

其中,Kdeg:受影响酶的表观一级降解速率常数(肝和肠分别使用了0.0005min-1),d:校正因子,设置为1. 肠道内促变药的浓度([I]g)使用以下公式计算:
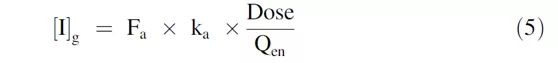
其中,Ka是一级吸收速率常数,Qen是肠上皮细胞的血流量(18 L/h)。肝脏中的促变药的浓度([I] h),肝脏入口处的最大未结合血浆的药物浓度,使用以下公式计算:
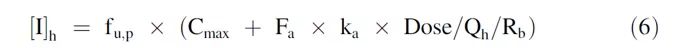
其中,fu,p:药物在血浆中的游离分数;Cmax:稳态时血浆中最大抑制剂总浓度(即游离部分加上结合部分);Qh:肝血流量(每70千克97 L/h);从口服5 mg Esaxerenone的浓度-时间曲线计算得出的Ka为1.19(1/h);fu,p和Rb为实测值;Cmax为87.2 ng/ml;使用未结合分数(fu, inc)将Ki,KI和EC50校正为未结合值。肝微粒体的游离药物分数(fu, inc)为体外实测值,肝细胞的游离药物分数(fu, inc)为GastroPlus预测值(根据Austin方程)。本研究计算了结合所有可用机制的AUCR(AUCRtot),还分别计算了抑制和诱导作用的AUCR(AUCRinh和AUCRind)、基于肠道和肝脏相互作用的AUCR(AUCRg和AUCRh)。
2.3使用动态PBPK模型的DDI风险评估
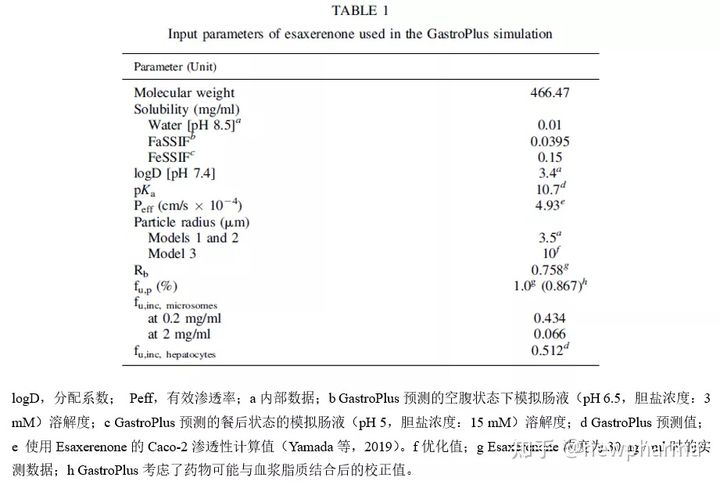
Esaxerenone PBPK模型的相关建模参数1). Esaxerenone PBPK模型输入的理化性质参数(表1)
2). Esaxerenone PBPK模型输入的处置参数(表2)
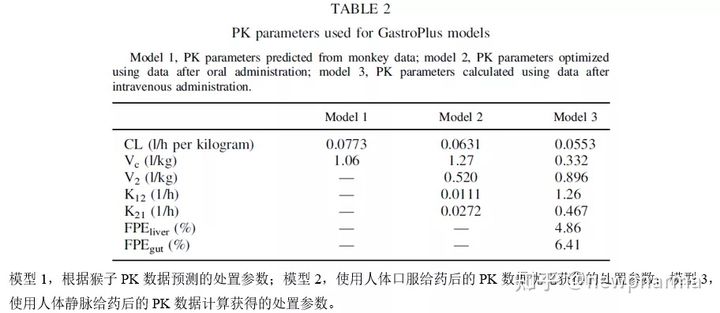
使用GastroPlus 进行了动态 DDI分析。其中胃肠道模型为高级隔室吸收和转运模型,表1和表2列出了用于Esaxerenone模型构建的参数。肠上皮细胞的未结合分数设置为100%。建立了三种具有不同处置学参数的Esaxerenone模型,以比较不同研发阶段DDI的可预测性。模型1,根据猴子静脉给药的PK数据,通过单物种异速缩放从猴子的清除率CL和分布体积Vd预测了人体的CL和Vd,计算方法如下:

为了评估可能产生的最大DDI风险,假定生物利用度为1,由于难以估算进出二房室的分配速率常数(即K12和K21),将模型1设定为1室模型。模型2为2-房室模型,其中CL, Vc,K12和K21为人体口服5mg Esaxerenone的PK数据的拟合值。模型3为2-房室模型,其中CL, Vc, K12和K21是使用静脉注射5mg Esaxerenone的PK数据计算得到,同时计算出首过效应(FPEs)并将其输入模型中(表2)。肝脏中的FPE由GastroPlus根据CL计算得出,假设吸收百分数为1,利用Esaxerenone的生物利用度(89.0%)计算得出Fg,结合Esaxerenone的口服PK数据对粒径做了优化,将颗粒半径从3.5μm增加到10μm。下表列举了作为底物的咪达唑仑PBPK模型的建模参数。3)咪达唑仑PBPK模型输入的理化与处置参数
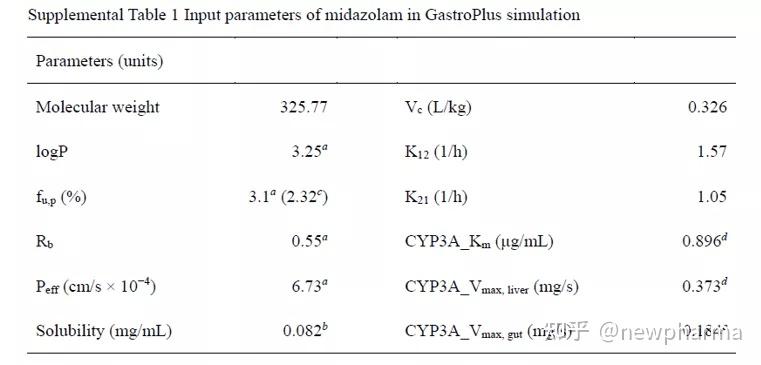
使用GastroPlus的DDI模块进行动态DDI模拟。除AUCR以外,还分别预测了抑制参数和诱导参数对应的AUCRinh和AUCRind. 其中Ki,KI和EC50与静态机制模型使用了相同数值;肠道和肝脏中CYP3A的Kdeg均使用0.0005min-1。计算了肠道利用率的比值AUCRg,肝脏利用率的比值AUCRh是通过AUCR除以AUCRg计算得到。在临床研究中,口服Esaxerenone 5 mg每天1次,持续14天,并在第14天同时口服咪达唑仑2 mg。在当前的研究中(模型3用于以下模拟),为模拟Esaxerenone多次给药后的作用持续时间,在第1、2、3和14天设置了咪达唑仑给药;为了模拟Esaxerenone和咪达唑仑不同间隔给药的DDI效果,在第14天服用Esaxerenone后,将咪达唑仑的给药时间分别设置为0、1、2和12小时。此外,还使用1.25mg至10 mg的Esaxerenone进行了DDI模拟。
3. 结果与分析
3.1体外DDI研究
1. Esaxerenone对人肝微粒体中P450和UGT活性的抑制能力。研究表明Esaxerenone抑制CYP2B6, CYP2C8, CYP2C9, CYP2C19和CYP3A, 对应的Ki值见表3。表3. Esaxerenone对CYP450酶的抑制作用:
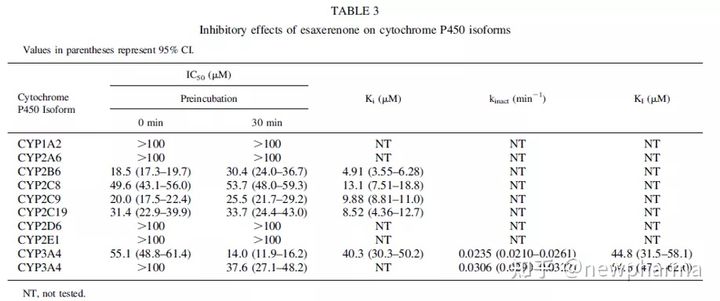
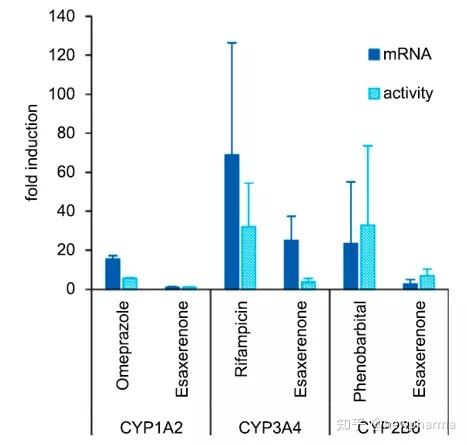
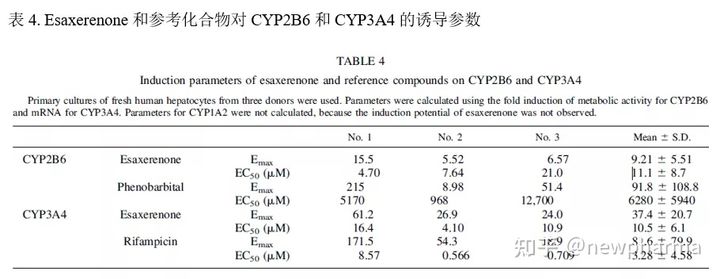
Esaxerenone不会对CYP1A2, CYP2A6, CYP2D6或CYP2E1的活性产生抑制,而对CYP3A4的活性具有时间依赖性抑制作用(参考IC50值,表3.)。并通过使用咪达唑仑和睾酮作为CYP3A4底物进行了灭活研究,获得了对应的Kinact和KI值,见表3. 使用人肝微粒体研究了Esaxerenone可逆地抑制UGT1A1和UGT2B7的能力。对应UGT1A1和UGT2B7的IC50值分别为23.6 mM和100 mM,对应UGT1A1的Ki为12.4 mM。Esaxerenone对人肝细胞中CYP450酶的诱导能力。考察了Esaxerenone诱导CYP1A2,CYP2B6或CYP3A4的能力。Esaxerenone没有表现出细胞毒性作用。比较了Esaxerenone与奥美拉唑、利福平或苯巴比妥在最大浓度下诱导CYP450酶的mRNA的表达和酶活性的倍数变化,如图2所示。Esaxerenone不诱导CYP1A2 mRNA表达或活性增加,但表现出对CYP2B6和CYP3A4的诱导作用。值得注意的是,可能是由于Esaxerenone对CYP3A4具有抑制作用,Esaxerenone引起的CYP3A4活性的增加小于mRNA表达的增加。鉴于mRNA通常比代谢活性更能指示诱导作用,因此使用mRNA数据计算了CYP3A4诱导的Emax和EC50。但对于CYP2B6,由于诱导CYP2B6的代谢活性的倍数比mRNA的倍数变化大,使用了代谢活性计算了诱导参数。表4列举了Esaxerenone和参考化合物(对于CYP3A4为利福平,对于CYP2B6为苯巴比妥)的对应计算值。Esaxerenone对CYP3A4诱导作用的Emax和EC50分别为24.0-61.2倍和为4.1-16.4 mM;对CYP2B6的诱导作用的Emax和EC50分别是5.52-15.5倍和4.70-21.0 mM。
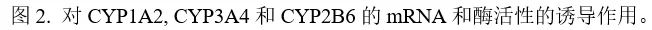
Esaxerenone对转运蛋白的抑制作用。结果表明Esaxerenone抑制OCT1的IC50为9.84 μM(3.79-15.9),OCT2为31.0 μM(24.2-37.7),MATE1为9.70 μM(6.78-12.6),MATE2-K为5.64 μM(4.38-6.91),BCRP为24.6 μM(4.81-44.5),P-gp为16.3 μM(12.2-20.5)。Esaxerenone抑制OAT1, OAT3, OATP1B1和OATP1B3的IC50值均超过30 μM。考虑到药物的全身暴露,OCT1, OCT2, MATE1和MATE2-K不太可能产生临床上的PK变化。关于P-gp和BCRP,由于Igut/ IC50分别为2.63和1.74,所以体内产生DDI的可能性较低(Igut/ IC50≥10,表示有DDI的可能)。
3.2使用静态机制模型的DDI风险评估结果
体外研究表明Esaxerenone抑制CYP2B6的Ki值最小,对CYP3A具有TDI。另外对CYP2B6和CYP3A4具有诱导作用。为评估Esaxerenone与CYP2B6或CYP3A4的底物进行临床DDI研究的必要性,本研究使用静态机制模型分析了可能的DDI程度。通过可逆抑制和诱导作用预测的CYP2B6酶活性变化分别为1.00(Ah)和1.02(Ch),这表明在临床中通过CYP2B6产生DDI的程度可忽略不计。由于CYP3A4在肠上皮细胞中的高表达,因此必须考虑肠道DDI的可能性。通过可逆抑制、TDI和诱导作用计算的肠道和肝脏中CYP3A4酶活性的倍数变化分别为0.961(Ag),0.099(Bg),5.76(Cg),1.00(Ah),0.946(Bh),1.03(Ch)。这表明肠道是Esaxerenone作为促变药通过TDI和诱导作用产生DDI的主要部位。当分别考虑抑制和诱导作用时,咪达唑仑的AUCRinh和AUCRind计算值分别为1.80和0.31。这表明尽管Esaxerenone的DDI作用不是很强,但仍不能忽略。抑制和诱导同时作用时计算的AUCR为1.30(根据CED和FDA发布的DDI指导原则,不建议使用静态机制模型估计抑制与诱导作用同时存在时的共同效应)。
3.3使用动态PBPK模型的DDI风险评估结果
建立了三个具有不同处置学参数的Esaxerenone模型,图3显示了在血浆和肠上皮细胞(以空肠1为例)中Esaxerenone的模拟浓度。尽管模型1和2模拟的血浆浓度不同(图3,A和B),但模拟肠细胞中的药物浓度(Cent)相同(图3,C和D),因此模拟AUCR也是相同,因为预测的Cent主要取决于溶解度、溶解速度和膜渗透性。对于模型3,输入了使用绝对生物利用度(89.0%)计算的FPE(肝脏为4.86%,肠道为6.41%)。由于使用实测颗粒半径预测的溶出度太高无法准确计算吸收曲线,因此通过优化颗粒半径对模型进行了校正,从而降低Cent。
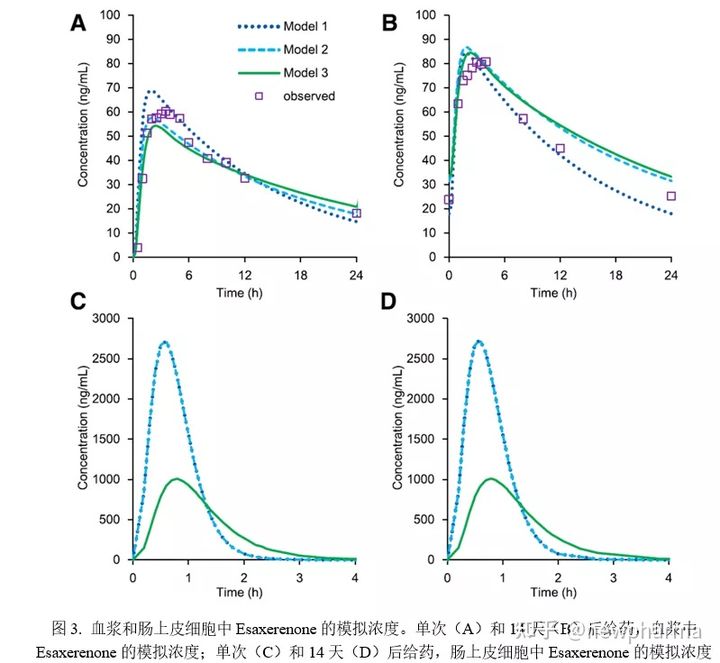
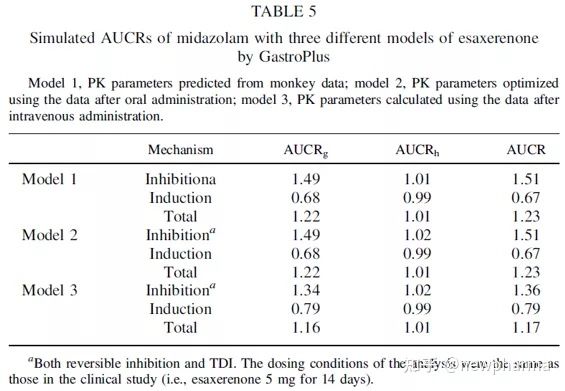
表5列举了与临床研究相同的给药方案,在仅存在抑制作用、诱导作用或同时二者同时存在时模型1-3所计算的肠道和肝脏中的AUCRs。尽管模型3的AUCRinh和AUCRind比模型1或2的略小,但AUCR与模型1或2相似。使用这三个模型计算出的AUCRtot范围为1.17-1.23,与临床研究实测值1.2接近,这表明Esaxerenone通过CYP3A介导的抑制和诱导作用产生DDI的程度较低。使用三个模型计算的AUCRh均约为1,即使仅单独考虑抑制或诱导作用也是如此,这表明相互作用主要发生在肠道中,而肝脏中相互作用较小,因为作为促变药的Esaxerenone相互作用力弱且临床暴露率低。
如图4所示,AUCRs是根据Esaxerenone的给药方案(第1、2、3和14天5 mg)、咪达唑仑的给药时间设置(即Esaxerenone给药后0、1、2或12小时)或者Esaxerenone剂量(即1.25、2.5、5或10 mg)计算得出的。单次口服5 mg Esaxerenone后咪达唑仑的AUCRinh小于14天的值,3天给药的AUCRinh略小于14天给药的AUCRinh,由于诱导比14天给药稍弱,因此第3天和第14天的AUCRtot几乎相同。与同时服用咪达唑仑和Esaxerenone相比,在Esaxerenone服用后1或2小时服用咪达唑仑会导致AUCRinh增大和AUCRind减小,但AUCRtot却相似。当在Esaxerenone给药12小时后服用咪达唑仑时,AUCRinh小于在Esaxerenone给药1或2小时后给药的AUCRinh,这可能是由于酶更新造成的。当使用不同剂量的Esaxerenone(即1.25-10 mg)进行DDI模拟时,AUCRinh升高而AUCRind降低,呈剂量依赖性。然而,由于抵消作用,AUCRtot几乎保持恒定,即由于抑制而引起的代谢酶活性的降低被诱导引起的酶表达量的增加所缓和。与静态机制模型的计算结果相比,使用以上模拟设置,PBPK模型模拟的抑制和诱导作用都更弱(即AUCRinh更小和AUCRind更大)。
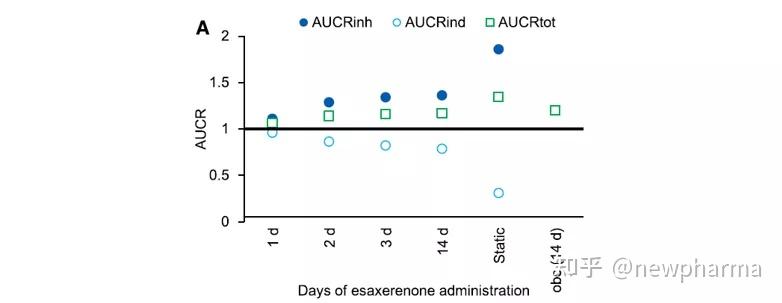
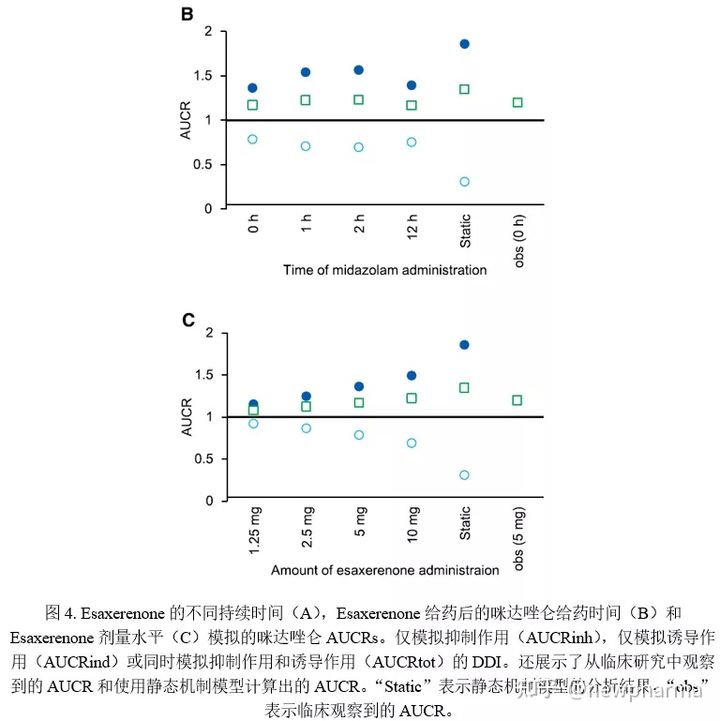
4 模型讨论
本研究通过评估体外、静态机制模型和动态PBPK模型评估了Esaxerenone作为促变药产生DDI的风险。讨论了PBPK建模软件GastroPlus用于预测肠道中弱DDI的实用性。根据静态机制模型分析,Esaxerenone表现出通过抑制主要酶或转运蛋白而发生临床DDI的风险很小或没有。尽管目前的结果表明Esaxerenone通过P-gp介导的DDI程度较低,但由于可能会与窄治疗窗药物地高辛合用,因此也进行了一项临床研究以考察Esaxerenone对地高辛(P-gp的典型底物)PK的影响。临床研究表明Esaxerenone对地高辛的稳态PK无影响,与静态机制模型分析结果一致。Esaxerenone作为的CYP3A的TDI和诱导剂需要进行模型分析以评估临床中DDI的风险。由于难以准确估计抑制和诱导之间的抵消效应,使用了静态机制模型分别考察了对CYP3A的TDI和诱导作用。此外,还进行了与咪达唑仑的临床DDI研究。在静态机制模型中假设使用了促变药的最大浓度,以避免造成假阴性,这种与真实世界不符的假设倾向于高估DDI风险,而可以模拟动态浓度变化的PBPK模型更可以实现对真实世界的预测。尽管通过PBPK建模分析CYP3A介导的DDI已广泛应用,但很多应用都集中在强相互作用,而对通过肠道CYP3A进行弱DDI的预测准确性尚不清楚。因此我们在先前的研究中使用17种市售药物证实了弱抑制性或抑制性和诱导性同时作用时的可预测性。为了比较使用有限数据搭建的模型对DDI的可预测性,考虑每个药物的实际开发过程,本研究搭建了三个用于动态模拟的模型。模型1是在进行首次人体研究之前,用猴子PK数据进行单种属异速缩放搭建了人体模型。模型2是在临床开发的早期阶段,只有口服给药的PK数据,并使用口服Esaxerenone后的PK数据搭建了动态PBPK模型,即使预测的处置学参数(CL, K12, K21等)与真实的参数之间存在差异,但也准确预测了肠道DDI。对于肠道DDI的预测,重要的是使用合理的溶解度、膜通透性,以及必要时要模拟肠内的新陈代谢,因此模型3考虑了FPE引起的Cent的降低,并且降低了溶出速度,然而诱导和抑制作用抵消后的AUCRtot值与模型1和2相当。根据之前及本研究的结果,对于CYP3A抑制和诱导作用均较弱的化合物,使用GastroPlus软件结合口服后的PK数据搭建的PBPK模型即可实现对DDI的准确预测。已有研究表明,当抑制和诱导同时发生时,所产生的DDI作用还和给药时间有关。为了了解这种复杂DDI的机理并指导临床研究设计,已有报道将PBPK模型用于相关研究。本研究模拟了不同剂量Esaxerenone与咪达唑仑的DDI,以及与咪达唑仑不同给药时间的DDI,但咪达唑仑的AUCRtot变化都很小,这是因为Esaxerenone对CYP3A的可逆抑制作用较弱,只有TDI和诱导作用会影响临床DDI。本研究还虚拟了可逆抑制和诱导作用都增强的情况下给药时间对AUCRtot的影响。考虑到Esaxerenone是用于治疗高血压的药物,它可能与其他Fg低于咪达唑仑的他汀类药物(即被肠道首过更多)合用。因此,本研究还模拟了Esaxerenone与这类虚拟底物的潜在DDI风险。
5 总结
本研究通过评估体外、静态机制模型和PBPK模型评估了Esaxerenone作为促变药产生DDI的风险。根据静态机制模型分析,Esaxerenone表现出通过抑制主要酶或转运蛋白而发生临床DDI的风险很小或没有。而Esaxerenone作为的CYP3A的TDI和诱导剂需要进一步进行模拟分析,以评估临床中DDI的风险。在静态机制模型中假设使用了促变药的最大浓度,以避免造成假阴性,这种与真实世界不符的假设倾向于高估DDI风险,因而基于PBPK模型进一步评估了DDI风险。考虑到药物开发不同阶段建模数据的完整性,本研究搭建了三个PBPK模型,以考察使用GastroPlus基于不同研发阶段的数据搭建的模型对肠道DDI的预测准确性。结果表明Esaxerenone虽然是CYP3A的TDI和诱导剂,但由于TDI作用和诱导作用的抵消效应,Esaxerenone作为促变药产生DDI的可能性较低,模拟结果与已有临床研究结果相近。此外,还模拟了Esaxerenone与咪达唑仑不同给药时间的DDI,以及不同剂量Esaxerenone与咪达唑仑的DDI。
6 应用软件与模块
该案例应用的软件是GastroPlus (version 9.7),涉及模块有Base, PKPlus, ADMET Predictor, Metabolism and Transporter Module, DDI Module。
参考文献
Yamada et al., Drug-Drug Interaction Risk Assessment of Esaxerenone as a Perpetrator by In Vitro Studies and Static and Physiologically Based Pharmacokinetic Models. Drug Metab Dispos , September 2020, 48:769–777.IF: 3.231
延伸阅读